KOKKOS with GPUs
Overview
Teaching: 15 min
Exercises: 15 minQuestions
How do I use KOKKOS together with a GPU?
Objectives
What is the performance like?
Using GPU acceleration through the KOKKOS package
In this episode, we shall learn to how to use GPU acceleration using the KOKKOS
package in LAMMPS. In a
previous episode, we
have learnt the basic syntax of the package command that
is used to invoke the KOKKOS package in a LAMMPS run. The main arguments and the
corresponding keywords were discussed briefly in that chapter. In this episode, we shall
do practical exercises to get further hands-on experiences on using those commands.
Command-line options to submit a KOKKOS GPU job in LAMMPS
In this episode, we’ll learn to use KOKKOS package with GPUs. As we have seen, to run the KOKKOS package the following three command-line switches are very important:
-k on: This enables KOKKOS at runtime-sf kk: This appends the “/kk” suffix to KOKKOS-supported LAMMPS styles-pk kokkos: This is used to modify the default package KOKKOS options
To invoke the GPU execution mode with KOKKOS, the -k on switch takes additional
arguments for hardware settings as shown below:
-k on g Ngpu: Using this switch you can specify the number of GPU devices,Ngpu, that you want to use per node.
Before you start
- Know your host: get the number of physical cores per node available to you.
- Know your device: know how many GPUs are available on your system and know how to ask for them from your resource manager (SLURM, etc.)
- CUDA-aware MPI: Check if you can use a CUDA-aware MPI runtime with your LAMMPS executable. If not then you will need to add
cuda/aware offto your<arguments>.
Creating a KOKKOS GPU job script
Create a job script to submit a LAMMPS job for the LJ system that you studied for the GPU package such that it invokes the KOKKOS GPU to
- accelerate the job using 1 node,
- uses all available GPU devices on the node,
- use the same amount of MPI ranks per node as there are GPUs, and
- uses the default package options.
Solution
#!/bin/bash -x # Ask for 1 nodes of resources for an MPI/GPU job for 5 minutes #SBATCH --account=ecam #SBATCH --nodes=1 #SBATCH --output=mpi-out.%j #SBATCH --error=mpi-err.%j #SBATCH --time=00:10:00 # Configure the GPU usage (we request to use all 4 GPUs on a node) #SBATCH --partition=develgpus #SBATCH --gres=gpu:4 # Use this many MPI tasks per node (maximum 24) #SBATCH --ntasks-per-node=4 #SBATCH --cpus-per-task=6 export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK export OMP_PROC_BIND=spread export OMP_PLACES=threads module purge module use /usr/local/software/jureca/OtherStages module load Stages/Devel-2019a module load intel-para/2019a # Note we are loading a different LAMMPS package module load LAMMPS/3Mar2020-gpukokkos srun lmp -in in.lj -k on g 4 -sf kk -pk kokkos cuda/aware offIf you run it how does the execution time compare to the times you have seen for the GPU package?
A few tips on gaining speedup from KOKKOS/GPU
This information is collected from the LAMMPS website
- Hardware comptibility: For better performance, you must use Kepler or later generations of GPUs.
- MPI tasks per GPU: You should use one MPI task per GPU because KOKKOS tries to run everything on the GPU, including the integrator and other fixes/computes. One may get better performance by assigning multiple MPI tasks per GPU if some styles used in the input script have not yet been KOKKOS-enabled.
- CUDA-aware MPI library: Using this can provide significant performance gain. If this is not available, set it
offusing the-pk kokkos cuda/aware offswitch.neighandnewton: For KOKKOS/GPU, the default isneigh = fullandnewton = off. For Maxwell and Kepler generations of GPUs, the default settings are typically the best. For Pascal generations, settingneigh = halfandnewton = onmight produce faster runs.- binsize: For many pair styles, setting the value of
binsizeto twice that used for the CPU styles could offer speedup (and this is the default for the KOKKOS/GPU style)- Avoid mixing KOKKOS and non-KOKKOS styles: In the LAMMPS input file, if you use styles that are not ported to use KOKKOS, you may experience a significant loss in performance. This performance penalty occurs because it causes the data to be copied back and forth from the CPU repeatedly.
In the following discussion, we’ll work on a few exercises to get familiarized on some of these aspects (to some extent).
Exercise: Performance penalty due to use of mixed styles
- First, let us take the input and job script for the LJ-system in the last exercise. Make a copy of the job script that uses the following additional settings for KOKKOS:
newton offneigh fullcomm deviceUse the number of MPI tasks that equals to the number of devices. Measure the performance of of this run in
timesteps/s.- Make a copy of the LJ-input file called
in.mod.ljand replace the line near the end of the file:thermo_style custom step time temp press pe ke etotal densitywith
compute 1 all coord/atom cutoff 2.5 compute 2 all reduce sum c_1 variable acn equal c_2/atoms thermo_style custom step time temp press pe ke etotal density v_acn- Using the same KOKKOS setting as before, and the identical number of GPU and MPI tasks as previously, run the job script using the new input file. Measure the performance of this run in
timesteps/sand compare the performance of these two runs. Comment on your observations.Solution
Taking an example from a HPC system with 2x12 cores per node and 4 GPUs, using 1 MPI task per GPU, the following was observed.
First, we ran with
in.lj. Second, we modified this input as mentioned above (to becomein.mod.lj) and performance for both of these runs are measured in units oftimesteps/s. We can get this information from the log/screen output files. The comparison of performance is given in this table:
Input Performance (timesteps/sec) Performance loss in.lj(all KOKKOS enabled styles used)8.097 in.mod.lj(non-KOKKOS style used:compute coord/atom)3.022 2.68 In
in.mod.ljwe have used styles that are not yet ported to KOKKOS. We can check this from the log/screen output files:(1) pair lj/cut/kk, perpetual attributes: full, newton off, kokkos_device pair build: full/bin/kk/device stencil: full/bin/3d bin: kk/device (2) compute coord/atom, occasional attributes: full, newton off pair build: full/bin/atomonly stencil: full/bin/3d bin: standardIn this case, the pair style is KOKKOS-enabled (
pair lj/cut/kk) while the compute stylecompute coord/atomis not. Whenever you make such a mix of KOKKOS and non-KOKKOS styles in the input of a KOKKOS run, it costs you dearly since this requires the data to be copied back to the host incurring a performance penalty.
We have already discussed that the primary aim of developing the KOKKOS package is to be able to write a single C++ code that will run on both devices (like GPU) and hosts (CPU) with or without multi-threading. Targeting portability without losing the functionality and the performance of a code is the primary objective of KOKKOS.
Performance comparison of CPU and GPU package (using KOKKOS)
Let us see now see how the current KOKKOS/GPU implementation within LAMMPS (version
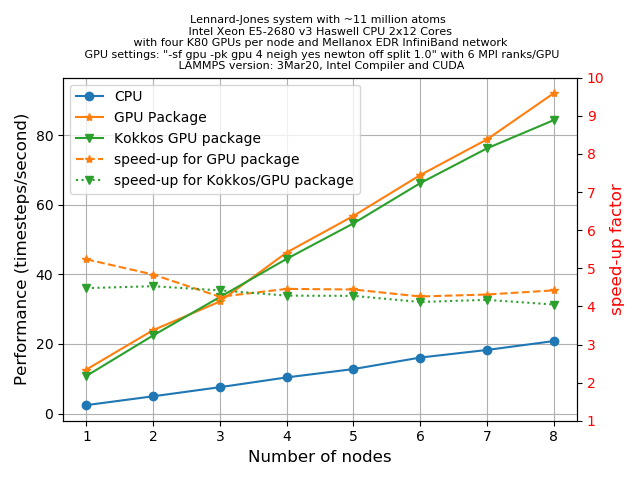
3Mar20) achieves this goal by comparing its performance with the CPU and GPU package. For this, we shall repeat the same set of tasks as described in episode 5. Take an LJ-system with ~11 million atoms by choosingx = y = z = 140andt = 500.KOKKOS/GPU is also specially designed to run everything on the GPUs (in this case there are 4 visible devices). We shall offload the entire force computation and neighbour list building to the GPUs using the
<arguments>:-k on g 4 -sf kk -pk kokkos newton off neigh full comm deviceor, if CUDA-aware MPI is not available to you,
-k on g 4 -sf kk -pk kokkos newton off neigh full comm device cuda/aware offWe have created a plot to compare the performance of the KOKKOS/GPU runs with the CPU runs (i.e. without any accelerator package) and the GPU runs (i.e. with the GPU package enabled) with various numbers of nodes:
Discuss the main observations you can make from this plots.
Solution
There is only marginal difference in the performance of the GPU and KOKKOS packages. The hardware portability provided by KOKKOS therefore make it an attractive package to become familiar with since it is actively maintained and developed and likely to work reasonably well on the full spectrum of available HPC architectures (ARM CPUs, AMD graphics cards,…) going forward.
A caveat on these results however, at the time of their generation mixed precision support in the LAMMPS KOKKOS package was still under development. When running large number of atoms per GPU, KOKKOS is likely faster than the GPU package when compiled for double precision. It is likely that there is additional benefit of using single or mixed precision with the GPU package (depending significantly on the hardware in use and the simulated system and pair style).
Key Points
Knowing the capabilities of your host, device and if you can use a CUDA-aware MPI runtime is required before starting a GPU run
KOKKOS compares very well with the GPU package in double precision
KOKKOS aims to be performance portable and is worth pursuing because of this